

Gene Sequencing and its Medical Applications
- Camille Rebouillat-Sarti

- Sep 26, 2022
- 5 min read
Since its emergence in the past few decades, gene sequencing has become indispensable in many areas of biology. The main sequencing methods available nowadays, including Sanger sequencing and Next-Generation Sequencing, are showing great promises, especially in the medical field (Krier et al., 2016). Among the most recent developments enabled by the main techniques aforementioned lie improved diagnosis through Whole Exome Sequencing, significant progress in cancer research, and even gene therapy for Spinal Muscular Atrophy. This article will therefore start by introducing the science behind the sequencing methods, examining the consequential improvements linked to them, before explaining some of the current challenges that these technologies are posing.
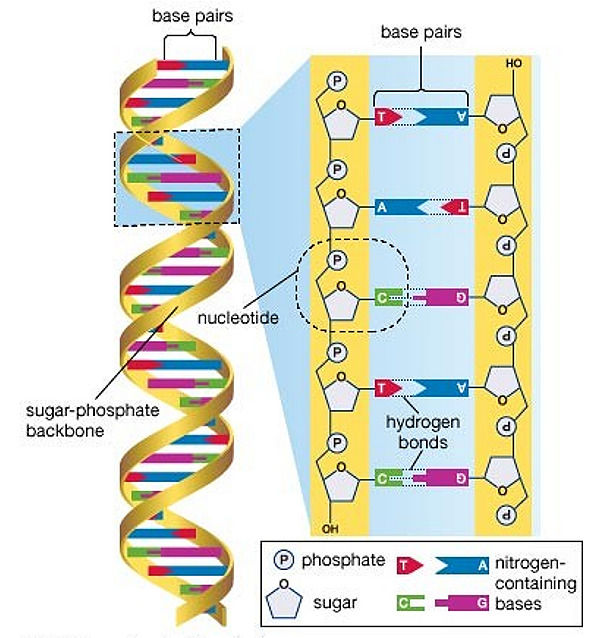
To start with, as Calladine explains in Understanding DNA, every human cell contains twenty-three pairs of chromosomes located in their nucleus, which are the structures that contain Deoxyribonucleic Acid (DNA). DNA is a large, complex molecule made of two anti-parallel strands held together by base-pair hydrogen bonds that wind into a double helix. These bases in the center, also known as nucleotides, are four in number: Adenine (A), Thymine (T), Cytosine (C) and Guanine (G). Adenine only pairs with Thymine, while Cytosine only pairs with Guanine. It is the sequence of these base-pairs that defines specific genes, and therefore encodes the synthesis of either Ribonucleic Acid (RNA) or proteins (Calladine et al., 2004). Gene sequencing – the process of determining the order of nucleotides that make up the DNA – thus gives access to all the genetic information required to build and maintain an organism, as well as to classify all the genes according to their location and function (Brown, 2017). This way, a reference genome can be constructed, which was the goal of the Human Genome Project launched in 1988 and completed in 2003 (Hood & Rowen, 2013).
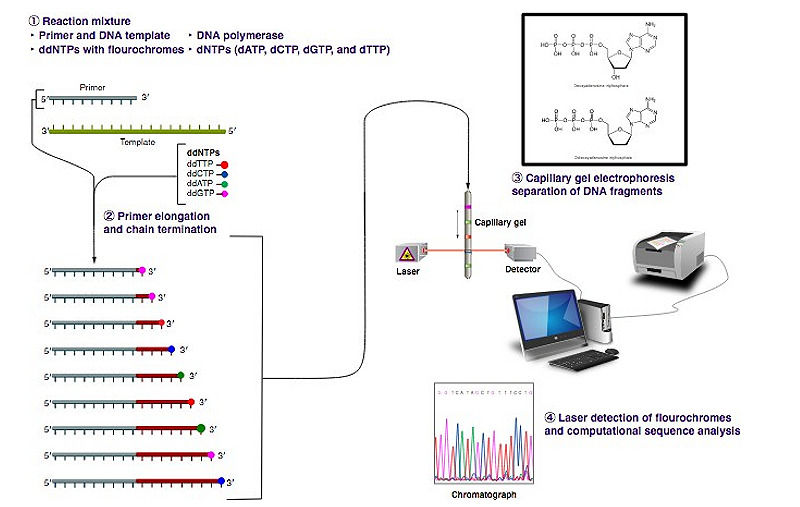
The whole human genome, however, comprises over three billion nucleotides, and therefore could not be sequenced all at once until recently, because most of the currently available techniques of DNA sequencing could not handle more than short stretches of DNA at a time (Nurk et al., 2022). The most common sequencing method is the Sanger method, or ‘chain-termination method’, developed by British chemist and Nobel Prize winner Frederick Sanger in the late 1970s (Brown, 2017). It consists in breaking the genome into smaller fragments of about 500 base-pairs, analyzing them individually, and then reconstructing the full sequence. The target DNA is copied many times, making clusters of identical DNA bits. Then, fluorescent chain-terminating nucleotides are incorporated in the process to mark the end of each fragment. Indeed, ’dideoxy’ versions of the four nucleotides with different color dyes are mixed with the fragments: these lack the hydroxyl group on the 3’ Carbon atom in the ring, preventing the adding of further nucleotides to the chain, while the dye molecule on the dideoxy-nucleotide is linked to the nitrogenous base (Brown, 2017). DNA is then copied again, and the resulting fragments go through electrophoresis: as smaller pieces travel quicker through the capillary gel, the ones that differ in length by only one base reach the sequencer in ascending order, where each dye is detected with a laser in the right order. Large-scale automatic sequencing machines finally translate the color sequence into an ATCG letter code (Brown, 2017).
Although Sanger sequencing is still widely used, it is more expensive and less efficient for larger-scale projects than the most recently developed sequencing methods collectively referred to as Next-Generation Sequencing (NGS). These high-throughput technologies consist in the massive parallelization of Sanger processes, enabling to sequence a much larger number of smaller strings of DNA and obtain much faster results at much cheaper costs (Brown, 2017). Whole Genome Sequencing (WGS) is now possible in just a few days, nevertheless the Whole Exome Sequencing (WES) approach is often privileged as it sequences only the exons – the protein-coding units of the genome. Several other NGS technologies have been developed since the 1990s which have dramatically reduced the cost of DNA sequencing as well as contributed largely to democratize its usage in biotechnology and biomedicine (Beale et al., 2015).
The advent of DNA sequencing has indeed greatly accelerated medical research and enabled new forms of diagnosis and treatment (Krier et al., 2016). The usual methods for detecting genetic disorders present multiple limitations. For instance, karyotyping, i.e., chromosome analysis, can only detect relatively large chromosomic abnormalities, while Comparative Genomic Hybridization, i.e., the 'detection of losses and gains in DNA copy number across the entire genetic genome without prior knowledge of specific chromosomal abnormalities' focuses only on unbalanced abnormalities that affect the DNA copy number (Beale et al., 2015, p.8). Unambiguous diagnosis is thus made difficult as many mutations may slip through the net if the latter are not performed in combination with other genetic tests. WES is currently revolutionising the field of genetic testing, allowing for a clear identification of specific genes responsible for certain disorders in one single test, and hence for an increased diagnostic yield (Beale et al., 2015). Moreover, the creation of cancer genomes that catalogue the genetic basis of different types of cancers is an ongoing topic of research (Upadhyay et al., 2014). NGS, especially WGS and WES, with their high-throughput and single-base resolution, enabled the ‘identification of variations underlying the cancer genomes, providing a comprehensive and high-quality set of common and rare polymorphisms and mutations’ (Upadhyay et al., 2014, p.797). Such knowledge of cancer-causing alterations gives hopes of discovering new, more targeted treatments in a close future.

Further, one of latest progress NGS enabled in the medical industry is the 2019 approval of Zolgensma® (OnasemnogeneAbeparvovec-xioi), a gene replacement therapy for the cure of infantile Spinal Muscular Atrophy (SMA) types I and II (Rosenmayr-Templeton, 2019). SMA is a genetic, hereditary disorder affecting 1 per 11,000 people worldwide (Kolb & Kissel, 2015). It is caused by a mutation in the SMN1 gene, that encodes the Survival Motor Neuron protein. Most patients are missing a piece of this gene, which prevents SMN protein production. As a result, the motor neurons die and the skeletal muscles atrophy. The patients can’t sit, stand, or walk without support, and most don’t survive past early childhood due to respiratory muscle weakness (Kolb & Kissel, 2015). However, most people are born with multiple ‘back-up’ SMN2 genes, that produce a small amount of functional SMN protein. The severity of the condition is thus often inversely proportional to the number of SMN2 copies in a person’s genome (Butchbach, 2016).
Genetic testing provides a precise diagnosis of which type of SMA is involved by identifying the SMN1 deletion/mutation and by counting the number of SMN2 copies (Cao et al., 2018). Nevertheless, not only does genetic sequencing enable to understand the causes of SMA, the Zolgensma® therapy showed outstanding results in early 2020. It is an adeno-associated AAV9 virus vector-based gene therapy, i.e., a recombinant form of the AAV9 virus carries one copy of the SMN1 transgene (instead of its own DNA) into the target motor neuron cells that will start replicating it, hence replacing the defective or missing gene (Rosenmayr-Templeton, 2019). Clinical trials hence 'enabled 19 out of the 21 children enrolled to survive significantly longer than would have been predicted based on the standard prognosis for children with this condition', Rosenmayr-Templeton (2019, p.3) explains.

Despite their potential, NGS technologies pose various technical and ethical challenges. Firstly, the colossal amount of data NGS provides is very difficult to store. Secondly, data analysis and interpretation remain controversial, and can only be achieved properly through a better understanding of the human genome altogether (Beale et al, 2015). Lastly, the adoption of genetic testing in health-care settings raises ethical, legal, and social issues, mainly addressing the questions of informed consent, privacy, genetic exceptionalism, and tremendous costs (Hodge, 2004). Besides, third-generation or ‘next-next generation’ sequencing is an ongoing research field, mainly focusing on single- molecular DNA sequencing, which would produce much longer reads and thus facilitate the reassembly of the genome transcript (Athanasopoulou et al., 2021). Routine gene sequencing owns therefore the potential to open crucial possibilities for biomedical applications, or biological research, bioinformatics, biotechnology, and forensics.
Bibliographical References
Athanasopoulou, K., Boti, M., Adamopoulos, P., Skourou, P., & Scorilas, A. (2021). Third-Generation Sequencing: The Spearhead towards the Radical Transformation of Modern Genomics. Life, 12 (1), 30. https://doi.org/10.3390/life12010030.
Beale, S., Sanderson, D., Sanniti, A., Dundar, Y., & Boland, A. (2015). A scoping study to explore the cost-effectiveness of next-generation sequencing compared with traditional genetic testing for the diagnosis of learning disabilities in children (pp. 7-14). Health Technology Assessment, No. 19.46.
Brown, T. (2017). Genomes 4 (4th ed.). Garland Science.
Butchbach, M. (2016). Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Frontiers In Molecular Biosciences, 3. https://doi.org/10.3389/fmolb.2016.00007.
Calladine, C., Luisi, B., Drew, H., & Travers, A. (2004). Understanding DNA - The Molecule & How It Works. https://doi.org/10.1016/b978-0-12-155089-9.x5000-5.
Cao, Y., Zhang, W., Qu, Y., Bai, J., Jin, Y., Wang, H., & Song, F. (2018). Diagnosis of Spinal Muscular Atrophy. Chinese Medical Journal, 131 (24), 2921-2929. https://doi.org/10.4103/0366-6999.247198.
Hodge, J. (2004). Ethical issues concerning genetic testing and screening in public health. American Journal Of Medical Genetics, 125C (1), 66-70. https://doi.org/10.1002/ajmg.c.30005.
Hood, L., & Rowen, L. (2013). The human genome project: big science transforms biology and medicine. Genome Medicine, 5 (9), 79. https://doi.org/10.1186/gm483.
Kolb, S., & Kissel, J. (2015). Spinal Muscular Atrophy. Neurologic Clinics, 33 (4), 831-846. https://doi.org/10.1016/j.ncl.2015.07.004.
Krier, J., Kalia, S., & Green, R. (2016). Genomic sequencing in clinical practice: applications, challenges, and opportunities. Dialogues In Clinical Neuroscience, 18 (3), 299-312. https://doi.org/10.31887/dcns.2016.18.3/jkrier.
Nurk, S., Koren, S., Rhie, A., Rautiainen, M., Bzikadze, A., & Mikheenko, A. et al. (2022). The complete sequence of a human genome. Science, 376 (6588), 44-53. https://doi.org/10.1126/science.abj6987.
Rosenmayr-Templeton, L. (2019). Industry update for May 2019. Therapeutic Delivery, 10 (9), 555-561. https://doi.org/10.4155/tde-2019-0043.
Upadhyay, P., Dwivedi, R., & Dutt, A. (2014). Applications of next-generation sequencing in cancer. Current Science, 107 (5), 795-802.
Visual Sources
Du Buisson, A. (2019). The Math That Tells Cells What They Are [Image]. Retrieved from: https://www.quantamagazine.org/the-math-that-tells-cells-what-they-are-20190313/.
Encyclpaedia Britannica, Inc. DNA Molecule [Image]. https://www.britannica.com/science/DNA-sequencing.
Estevezj. (2012). The Sanger (chain-termination) method for DNA sequencing [Image]. https://www.khanacademy.org/science/high-school-biology/hs-molecular-genetics/hs-biotechnology/a/dna-sequencing.
Padgett, C. (2016) DNA-Sequencing Research. [Image]. https://www.scientificamerican.com/article/why-gene-tests-for-cancer-don-t-offer-more-answers/.
Spinal Muscular Atrophy UK. Figure adapted from Burghes, A.H. and Beattie, C.E., Burghes, A.H. and Beattie, C.E. (2009) ‘Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?’ Nature Reviews Neuroscience, 10, pp. 597-609. [Image]. https://smauk.org.uk/the-genetics-of-5q-sma.





With professionalism and a great writing, the writer managed to make a difficult topic easy to understand.